Informaţii generale şi recomandări pentru efectuarea testului genetic
Fibroza chistică sau mucoviscidoza este cea mai frecventă afecţiune monogenică, având o incidenţă de 1:2500-3500 nou-născuţi în rândul populaţiei de origine caucaziană. Se transmite autozomal recesiv şi se caracterizează prin pleiomorfism clinic, cu evoluţie cronică, având o durată medie de supravieţuire de 37 ani, vârstă ce este mai mare la bărbaţi decât la femei.
|
Rasă sau grup etnic |
Frecvenţa purtătorilor (heterozigoţilor) |
Rata de detecţie a mutaţiilor |
|
Americani de origine africană |
1/65 |
77% |
|
Evreii Ashkenazi |
1/25 |
99% |
|
Americani de origine asiatică |
1/90 |
54% |
|
Europeni |
1/25 |
80% |
|
Est europeni |
1/25 |
75% |
|
Români |
1/30 |
65% |
|
Canadieni de origine franceză |
1/25 |
91% |
|
Americani de origine hispanică |
1/46 |
81% |
|
Nord europeni |
1/25 |
91% |
|
Sud europeni (italieni) |
1/25 |
77%5 |
Boala este cauzată de mutaţii la nivelul unei genei situate pe braţul lung al cromozomului 7 (7q31), care codifică o proteină transmembranară, CFTR (Cystic Fibrosis Transmembrane Conductance Regulator), ce face parte din familia proteinelor cu activitate ATP-azică şi funcţionează ca un canal de clor la nivelul polului apical al membranei celulelor epiteliale. În plus, proteina este implicată şi în reglarea canalelor de sodiu, intervine în transportul HCO3– prin membranele celulelor epiteliale şi poate acţiona ca un canal pentru alte proteine, cum ar fi glutationul. Studii proteomice recente au demonstrat că CFTR interacţionează cu multe proteine intracelulare, însă relevanţa fiziopatologică a acestor interacţiuni nu a fost încă pe deplin elucidată1.
După depistarea în 1989 a defectului genetic de la nivelul genei implicate în fibroza chistică, s-a crezut că un număr limitat de mutaţii cauzează această boală, însă până în prezent au fost descrise mai mult de 1500 mutaţii diferite. Aproape toate sunt mutaţii punctiforme sau deleţii mici (de la 1 la 84 bp). Cu toate acestea, este important să se înţeleagă că majoritatea sunt rare, iar consecinţele funcţionale ale multora dintre ele sunt greu de înţeles. De fapt mai puţin de 10 mutaţii (evidenţiate în tabelul 2), apar cu o frecvenţă mai mare de 1%, în timp ce cea mai frecventă mutaţie la nivel mondial, caracterizată prin deleţia fenilalaninei în poziţia 508 (ΔF508 – Phe508del) (deleţia a 3 perechi de baze de la nivelul exonului 10), se găseşte la aproximativ 30-80% dintre pacienţii cu mucoviscidoză, în funcţie de grupul etnic afectat. În tabelul 1 sunt enumerate 38 dintre mutaţiile testate în laboratorul nostru pentru stabilirea diagnosticului de fibroză chistică sau testarea purtătorilor1;3.
Mutaţiile genei CFTR pot fi grupate în 6 clase diferite, împărţite în raport cu consecinţele lor funcţionale la nivel celular (vezi figura 18.1.3.3.1):
- clasa I: proteina nu este sintetizată;
- clasa II: CFTR este insuficient prelucrată la nivelul aparatului Golgi;
- clasa III: proteina nu este funcţională;
- clasa IV: CFTR prezintă conductanţă anormală;
- clasa V: CFTR prezintă un defect parţial de sinteză;
- clasa VI: CFTR este degradată accelerat1.
Mutaţiile din clasele I, II, III sunt mai frecvente şi se asociază cu insuficienţă pancreatică, în timp ce mutaţiile din clasele IV, V şi VI sunt mai rare, iar pacienţii nu prezintă manifestări pancreatice1.
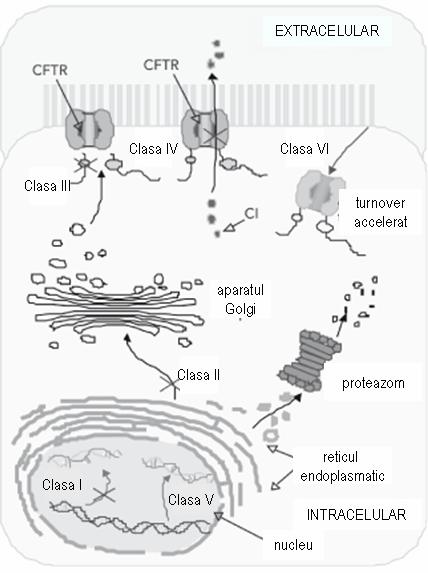
Figura 18.1.3.3.1 Adaptare după Roberta Rodrigues; Carmen S. Gabetta, Karla P. Pedro, Fabio Valdetaro, Maria I. M. Fernandes, Patrícia K. R. Magalhães, José N. Januário, Léa M. Z. MacielI, Cystic fibrosis and neonatal screening, Cad. Saúde Pública vol. 24 suppl.4, 2008.
Tabelul 1
|
F508del |
I507del |
F508C |
I502T |
1677del TA |
G542X |
|
1717-1G>A |
R553X |
G551D |
S549R A>C |
N1303K |
4016insT |
|
R1158X |
W1282X |
G1244E |
2789+5G>A |
711+5G>A |
711+1G>T |
|
G85E |
3849+10kbC>T |
R117H |
D1152H |
L1065P |
R1066H |
|
4382delA |
1259insA |
852del 22 |
R347P |
S912X |
I148T |
|
3199del6 |
1706del17 |
Q552X |
R1162X |
2183AA>G |
621+1G>T |
|
T338I |
L1077P |
Tabelul 2
|
Mutaţii |
Frecvenţa |
Clasa |
Fenotipul |
|
ΔF508 |
66.0% |
II |
Clasic |
|
G542X |
2.4% |
I |
|
|
G551D |
1.6% |
III |
|
|
N1303Lys |
1.3% |
II |
|
|
W1282X |
1.2% |
I |
|
|
R553X |
0.7% |
I |
|
|
621+1G>T |
0.7% |
I |
|
|
1717-1G>A |
0.6% |
I |
|
|
R117H |
0.3% |
IV |
Non-clasic |
|
R1162X |
0.3% |
Nedeterminată* |
Clasic6 |
* Transcripţia se realizează în condiţii normale, dar proteina este trunchiată, probabil incorect împachetată, de aceea cel mai probabil face parte din clasa II.
Gena CFTR conţine aproximativ 250-280 kilobaze, fiind alcătuită din 27 exoni. Aceasta codifică o glicoproteină constituită din 1480 aminoacizi, ce formează cinci domenii: 2 domenii transmembranare, fiecare cu 6 deschideri în α-helix, 2 domenii nucleotidice (NBD) în citoplasmă, interconectate la regiunile transmembranare şi un domeniu reglator (R), care leagă între ele domeniile transmembranare. Canalul ionic se deschide doar atunci când regiunea reglatoare a fost fosforilată de către proteinkinaza A (PKA), iar domeniul nuceotidic leagă ATP (vezi fig.18.1.3.3.2).
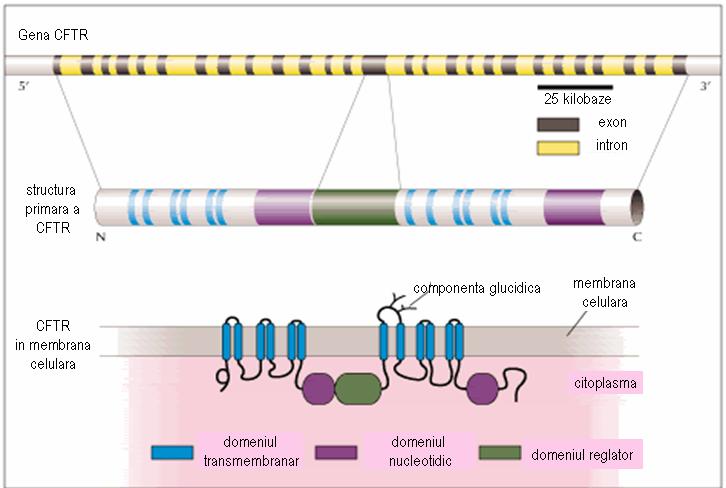
Figura 18.1.3.3.2 Gena CFTR (Adaptare după http://www.chromosome7.htmlplanet.com/custom4.html)
Consecinţa anomaliilor genetice este reprezentată de absenţa sau funcţionarea inadecvată a canalelor de clor la nivel celular, ceea ce se traduce prin alterarea transportului clorurilor în glandele mucoase şi seroase de la nivelul majorităţii organelor. Aceste secreţii vor avea un conţinut scăzut de apă, vor fi vâscoase, aderente la epiteliile ductelor excretoare şi greu de eliminat spre exterior. Acumularea acestora produce în timp alterarea funcţiilor şi distrucţia diferitelor organe (plămâni, pancreas, ficat, intestin, organe de reproducere). La nivelul pielii apare o secreţie sudorală cu concentraţie ridicată de sare.
Mucoviscidoza variază ca severitate în funcţie de mutaţiile CFTR şi factorii de mediu şi se prezintă sub mai multe forme, unele care determină moartea precoce a copiilor, ca urmare a unei boli pulmonare obstructive progresive cu bronşiectazii, altele caracterizate prin insuficienţă pancreatică şi boală pulmonară obstructivă progresivă în timpul adolescenţei cu creşterea frecvenţei de spitalizare la maturitate, iar altele manifestate prin sinuzite şi bronşite recurente sau infertilitate la bărbaţii tineri.
Prezentarea clinică, vârsta la diagnostic, severitatea simptomelor şi rata de progresie a bolii în organele implicate variază pe scară largă1;2;6.
Manifestări pulmonare. In utero, la naştere şi imediat după aceea, copiii cu fibroză chistică prezintă histologie normală a plămânilor (cu excepţia ductelor glandelor submucoase din căile respiratorii care sunt dilatate). La scurt timp însă, apar modificările pulmonare, ce se caracterizează prin obstrucţia căilor aeriene periferice ca urmare a acumulării de secreţii. Persoanele afectate dezvoltă ulterior un sindrom inflamator la nivelul tractului respirator inferior şi apoi infecţii cronice endobronşice. Deoarece elastaza (NE) eliberată de neutrofile la locul inflamaţiei determină clivajul receptorului CR1, a componentei C3b a complementului şi a imunoglobulinei G (IgG), bacteriile nu mai pot fi opsonizate şi distruse fiind astfel favorizată persistenţa acestora. Alterarea apărarii antiinfecţioase duce la apariţia bronşitelor şi bronşiolitelor bacteriene (cel mai frecvent cu Staphylococcus aureus şi Pseudomonas aeruginosa, dar şi Aspergillus fumigatus cu dezvoltarea de micetoame), cu infiltraţie neutrofilică intensă şi obstrucţia tractului respirator. Prezenţa elastazei determină de asemenea creşterea la nivelul celulelor epiteliale a producţiei de IL-8, care este chemoatractant pentru neutrofile, distruge elastina şi acţionează ca secretagog, contribuind la menţinerea inflamaţiei şi infecţiei şi producând astfel leziuni structurale şi tulburări ale schimbului de gaze2.
Simptomele respiratorii pot începe din prima lună de viaţă cu tuse, tahipnee sau wheezing, unii copii putând prezenta chiar detresă respiratorie severă asociată cu bronşiolită.
Deoarece polipii nazali apar la 10-32% dintre pacienţii cu fibroză chistică, prezenţa acestora reprezintă o indicaţie pentru efectuarea testului sudorii. Majoritatea copiilor de peste 8 luni (90%) prezintă sinuzită recurentă refractară la terapia cu antibiotice.
La adulţi, manifestările mucoviscidozei se caracterizează prin tuse cronică (la început uscată, intermitentă, concordantă cu episoadele infecţioase, ulterior se prelungeşte în timp, devine paroxistică, cu exacerbări nocturne şi în special dimineaţa la trezire, apoi productivă), expectoraţie intermitentă cu creşterea producţiei de spută asociată cu modificări ale culorii acesteia, febră uşoară, dispnee de efort, pneumonie, bronşiolită recurentă, atelectazie, hiperreactivitatea căilor respiratorii cu apariţia astmului refractar şi lipsa răspunsului la tratamentul cu agenţi β-adrenergici, bronşiectazii, hemoptizie, pneumotorax şi retracţii suprasternale şi intercostale. Pe măsură ce boala pulmonară progresează, ca urmare a infecţiilor cronice, apar leziuni structurale importante ale căilor respiratorii (chisturi sau abcese) însoţite de fibroza parenchimului pulmonar adiacent şi apariţia obstrucţiei cu hiperinflaţie şi blocarea gazelor. Hipoxemia apare iniţial în timpul somnului şi efortului fizic, iar hipercapnia se dezvoltă tardiv şi este un element de prognostic negativ. De asemenea, apariţia hipertensiunii pulmonare şi dezvoltarea cordului pulmonar odată cu boala parenchimatoasă avansată reprezintă un semn de prognostic nefavorabil, care este asociat cu o supravieţuire de aproximativ 8 luni.
Un studiu recent a raportat că expunerea pasivă la fumul de ţigară afectează negativ funcţia pulmonară la persoanele cu fibroză chistică.
În ciuda tuturor intervenţiilor terapeutice, bolile pulmonare rămân cauza majoră de morbiditate şi mortalitate în fibroza chistică2;3;5;6.
Manifestări gastrointestinale. Mutaţiile de la nivelul genei CFTR determină apariţia unei secreţii alterate de clorură şi apă la nivel intestinal, ce poate conduce la apariţia ileusului meconial la naştere (mai frecvent la homozigoţii ΔF508) sau a sindromului de obstrucţie intestinală distală mai târziu în viaţă.
Ileusul meconial apare la 15-20% din nou-născuţii diagnosticaţi cu mucoviscidoză, putând fi principala manifestare în perioada neonatală. Sindromul de impactare meconială este determinat de acumularea de masă mucofecaloidă lipicioasă în ileonul terminal şi cec, aderentă la peretele intestinal, ce uneori se poate calcifica.
Atrezia jejunală asociată frecvent cu volvulus şi peritonită meconială poate fi de asemenea o formă de prezentare a fibrozei chistice.
Diagnosticul de mucoviscidoză poate fi suspectat încă din viaţa intrauterină datorită evidenţelor ultrasonografice sugestive pentru obstrucţie intestinală, cum ar fi aspectul hiperecogen al intestinului fetal.
Cauza sindromului de obstrucţie intestinală distală este neclară, putându-se asocia cu deshidratarea, febra, reducerea suplimentelor enzimatice, boala hepatică sau utilizarea medicamentelor cu efect antiperistaltic (opiacee, anticolinergice). Clinic se caracterizează prin crampe recurente, prezenţa unei mase palpabile la nivelul abdomenului şi semne de obstrucţie intestinală completă sau parţială. 50% dintre aceşti pacienţi suferă complicaţii cum ar fi peritonită, volvulus, atrezie, necroză, perforaţie sau formarea unui pseudochist.
Impactarea mucoidă a apendicelui poate fi observată ca o masă palpabilă asimptomatică în cadranul abdominal inferior.
Ca urmare a creşterii presiunii intraabdominale secundare tusei, aceşti pacienţi dezvoltă frecvent prolaps rectal.
Colonopatia fibrozantă este o entitate caracterizată prin strictura colonului ascendent, ce a fost descrisă iniţial în 1994 şi este caracteristică fibrozei chistice. Dozele mari de enzime pancreatice, în special lipaze, au fost incriminate în etiologia bolii. Simptomele iniţiale sunt similare cu cele din sindromul obstructiv, iar diagnosticul se stabileşte în urma efectuării unui tranzit baritat.
Incidenţa tumorilor gastrointestinale (esofagiene, gastrice, intestinale, hepatice, biliare, pancreatice şi retroperitoneale) este în creştere la pacienţii cu fibroză chistică, colonul fiind cea mai frecventă localizare a cancerului.
Insuficienţa pancreatică exocrină (cauzată de obstrucţia ductelor pancreatice cu secreţii, apariţia autodigestiei enzimatice şi în final a fibrozei interstiţiale) însoţită de sindrom de malabsorbţie apare încă de la naştere la marea majoritate a persoanelor diagnosticate cu FC, cu excepţia celor care au anumite mutaţii CFTR şi nu necesită administrarea de enzime pancreatice. Aceştia riscă totuşi să dezvolte pancreatită acută sau recurentă.
Manifestările clinice sunt steatoreea, distensia abdominală, durerile abdominale, flatulenţa, retardul de creştere (determinat de sindromul de malabsorbţie şi anemia hemolitică), defectele de coagulare sau erupţiile cutanate asociate cu deficienţele de vitamine liposolubile (A, D, E şi K) şi zinc. La aceşti pacienţi se recomandă administrarea de enzime pancreatice. Persoanele cu FC şi funcţie pancreatică nomală au o evoluţie clinică mai blândă cu rata medie de supravieţuire mai mare (56 ani) faţă de cei cu insuficienţă pancreatică.
La copii, deficitul de vitamine, electroliţi şi proteine se manifestă prin: fontanele proeminente; anemie hemolitică prin deficitul de vitamină E; complicaţii hemoragice ce pot rezulta prin deficitul de vitamină K datorat insuficienţei hepatice sau malabsorbţiei; hipoproteinemie şi edeme; hepatomegalie însoţită de creşterea valorilor enzimelor hepatice; acrodermatită enteropatică, constipaţie, deshidratare hiponatremică/hipocloremică şi piele sărată, alcaloză metabolică hipokaliemică secundară pierderii cronice de sare.
Scăderea toleranţei la glucoză este frecventă la bolnavii cu mucoviscidoză (în medie 40%). Diabetul zaharat asociat cu fibroza chistică (DZAMV) se manifestă în adolescenţă (prin cetoacidoză şi cetonurie), fiind diagnosticat la 7% din pacienţii cu vârsta între 11 şi 17 ani, cu creşterea prevalenţei la maturitate. Este mai frecvent la homozigoţii ΔF508 şi la sexul feminin. DZAMV reprezintă o entitate distinctă faţă de diabetul de tip 1 sau 2, dar are caracteristici comune cu ambele tipuri. Etiologia este o combinaţie între secreţia redusă de insulină (secundară fibrozei pancreatice şi numărului redus de celule insulare) şi rezistenţa periferică la insulină. Apariţia acestui tip de diabet se asociază cu un declin al funcţiei pulmonare, cu o stare nutriţională mai precară şi cu supravieţuire mai scurtă.
Odată cu creşterea perioadei de supravieţuire la aceşti pacienţi, bolile hepatobiliare asociate mucoviscidozei au devenit o complicaţie serioasă şi frecventă ce pot afecta calitatea vieţii pacienţilor. Implicarea hepatobiliară devine clinic aparentă după primii 10 ani de viaţă.
Absenţa proteinei CFTR funcţională în celulele epiteliale care tapetează ductele biliare determină formarea unei secreţii vâscoase, care obstruează canaliculele. Dacă acest proces este extins apare ciroză obstructivă, ce se poate complica cu varice esofagiene, splenomegalie şi hipersplenism.
Nou-născuţii cu fibroză chistică pot prezenta icter obstructiv prelungit prin stază biliară intra şi extrahepatică.
Calculii biliari au prevalenţă mai mare la pacienţii cu mucoviscidoză (15% dintre adulţii tineri cu fibroză chistică) decât la subiecţii normali.
Boala hepatică este a doua cauză de mortalitate după afecţiunile pulmonare în fibroza chistică2;3;5;6.
Fertilitate. Mai mult de 95% dintre bărbaţii cu FC sunt infertili, ca urmare a absenţei congenitale bilaterale a ductelor deferente şi mai rar prin azoospermie obstructivă. Corpul, coada epididimului şi veziculele seminale poate fi anormal dilatate sau chiar absente. Doar 1% dintre pacienţii cu mucoviscidoză sunt fertili, aceştia prezentând forme uşoare de boală. Spermatogeneza este normală la aceşti barbaţi.
Femeile cu FC sunt fertile, deşi câteva (20%) pot prezenta un mucus cervical anormal care contribuie la infertilitate.
Deoarece supravieţuirea indivizilor cu FC s-a îmbunătăţit considerabil în ultimele decenii, sarcina la femeile cu FC a devenit o problemă importantă. Există studii care afirmă că sarcina decurge bine la aceste pacientele, dar şi studii care atestă apariţia unor complicaţii.
Atât pentru mamă cât şi pentru făt, factorii de predicţie negativi în cazul unei sarcini sunt capacitatea vitală forţată (CVF) mai mică de 50% din valoarea prezisă şi starea nutriţională proastă. De fapt, o valoare a CVF mai mică de 50% din valoarea prezisă reprezintă o contraindicaţie absolută a sarcinii.
Cu toate acestea, tratamentul adecvat al afecţiunilor pulmonare, managementul agresiv al infecţiilor cu o mare varietate de antibiotice şi îmbunătăţirea nutriţiei au făcut ca sarcina să fie bine tolerată, în special la femeile cu forme uşoare sau moderate. La acestea factorii de risc pentru deteriorarea sănătăţii şi deces precoce după sarcină sunt aceiaşi ca şi pentru populaţia adultă non-gravidă. Într-un studiu recent, Goss a luat în considerare valoarea VEF (volumul expirator forţat), greutatea, înălţimea şi rata de exacerbare pulmonară pe an şi a constatat că sarcina nu a fost asociată cu un risc crescut de deces. De fapt, sarcina nu pare să prezinte riscuri suplimentare nici pentru subgrupurile de femei cu diabet zaharat sau cu valoare FEV mai mică de 40% din valoarea prezisă. Factorii predictori importanţi pe parcursul sarcinii sunt severitatea insuficienţei pulmonare materne şi starea de nutriţie, în sensul că deteriorarea acestora pot precipita naştere prematură. În ciuda tuturor eforturilor rata de nou-născuţi vii la femeile cu mucoviscidoză cu vârste cuprinse între 13 şi 45 ani este de 1.9%.
Riscul de apariţie a anomalilor congenitale la făt nu este crescut, iar alăptatul nu reprezintă o contraindicaţie2;3;6.
Manifestări osteoarticulare. Osteoartropatia hipertrofică afectează peste 6% dintre bolnavi, fiind mai rară la copiii mici şi mai frecventă la sexul masculin (2:1). Este un sindrom caracterizat prin proliferare anormală a ţesutului osos la nivelul extremităţilor distale ale oaselor lungi, cu apariţia hipocratismului digital, durerii sau tumefacţiei articulare, de obicei simetrică, şi a periostitei.
Artropatia în fibroza chistică apare la 10% dintre bolnavi, putându-se manifesta în orice moment evolutiv. Etiologic sunt implicate mecanisme imunologice probabil legate de infecţia cronică cu Pseudomonas aeruginosa. Debutul este brusc cu durere, tumefacţie şi uneori se poate asocia cu leziuni tegumentare cum ar fi rash-ul maculopapular, eritemul nodos şi ocazional purpura. Poate fi mono- sau poliarticulară, afectând articulaţiile mari sau mici, cele mai frecvente fiind genunchiul, glezna, articulaţia mâinii, cotului sau umărului.
Osteopenia sau osteoporoza sunt manifestări frecvente ale pacienţii cu FC şi sunt asociate cu apariţia fracturilor şi a cifozei3.
Clasic, diagnosticul fibrozei chistice se stabileşte pe baza elementelor clinico-anamnestice caracteristice şi este apoi confirmat prin testul sudorii sau analiza moleculară.
La 70% dintre pacienţi diagnosticul este stabilit înaintea vârstei de 1 an, de obicei în primele luni de viaţă. Totuşi există bolnavi la care diagnosticul este confirmat abia după vârsta de 10 ani.
Diagnosticul de FC poate fi stabilit la persoanele suspicionate dacă prezintă:
1. una sau mai multe caracteristici fenotipice ale FC;
şi
2. dovada anomaliei funcţiei CFTR:
- prezenţa mutaţiilor cauzatoare de boală în gena CFTR;
sau
- 2 valori anormale ale clorurilor în sudoare la iontoforeza cantitativă pilocarpinică (> 60 mEq/L);
sau
- valori specifice ale diferenţei de potenţial nazal.
Testul sudorii rămâne gold standard-ul în diagnosticarea bolii şi evaluează concentraţia de ioni de clor şi sodiu din transpiraţie. Valorile normale ale electroliţilor în sudoare se situează <40mMol/L; valorile pozitive sunt la copii >60 mMol/L, iar la adolescenţi şi adulţi tineri >70 mMol/lL; valorile echivoce: între 40-60 mMol/L se repetă obligatoriu şi se interpreteză în context clinic. O concentraţie de clorură de mai mare de 60 mMol/L în sudoare, la două determinări diferite stabileşte diagnosticul de boală.
Rezultatele fals pozitive pot fi asociate cu sindromul Hurler, iar cele fals negative pot să apară în cazul pierderilor acute de sare. În cazul în care FC este suspectată la un individ cu hiponatremie şi hipocloremie, testul sudorii ar trebui amânat până la restabilirea unui echilibrul electrolitic3;6.
În următoarele situaţii speciale testarea genetică este testul iniţial de diagnostic:
- diagnosticarea in utero a feţilor cu risc ridicat (în 2002, 4% din persoanele nou diagnosticate au fost identificate cu ajutorul diagnosticului prenatal);
- testarea prenatală a feţilor cu risc scăzut, dar cu imagini ecografice sugestive pentru boală;
- screening-ul nou-născuţilor (în 2002, 12.8% din persoanele nou diagnosticate au fost identificate prin intermediul screening-ului neonatal);
- testarea sugarilor simptomatici (cu ileus meconial), care sunt prea mici pentru a produce un volum adecvat de sudoare;
- testarea unei persoane simptomatice care are rude cu mutaţii CFTR identificate6.
Deoarece mucoviscidoza se transmite autozomal recesiv, în momentul concepţiei fiecare frate al unui individ afectat are 25% şanse să fie purtător şi să prezinte afecţiunea, 50% şanse să fie purtător asimptomatic şi 25% şanse să fie nepurtator şi să nu fie afectat.
Testarea prenatală se efectueză pe celule fetale obţinute prin biopsia vilozităţilor coriale prelevate la aproximativ 10-12 săptămâni de gestaţie sau prin amniocenteză, de obicei la circa 15-18 săptămâni de viaţă intrauterină.
Testul sudorii trebuie efectuat postnatal la toţi pacienţii la care fibroza chistică a fost suspectată.
Testarea genetică are un rol important în detectarea unor mutaţii cu implicaţii importante în determinismul anumitor fenotipuri. Cea mai bună corelaţie între genotip şi fenotip este legată de funcţia pancreatică. Mutaţiile cele mai frecvente au fost clasificate în două categorii: cele care determină insuficienţă pancreatică şi cele care se asociază cu funcţie pancreatică normală (denumite „pancreatic sufficient”, PS). Persoanele fără afectare pancreatică au, de obicei, una sau două alele mutante de tip PS, ce sunt dominante în ceea ce priveşte fenotipul pancreatic.
În schimb, corelaţia genotip-fenotip este, în general, slabă pentru bolile pulmonare în fibroza chistică. Afecţiunile pulmonare în rândul persoanelor cu genotipuri identice variază foarte mult, o explicaţie plauzibilă fiind intervenţia factorilor de mediu.
Severitatea bolii pulmonare la persoanele cu una sau două mutaţii R117H depinde de lungimea regiunii poli T de la nivelul intronului 8, astfel dacă bolnavul prezintă varianta 5T în configuraţie cis dezvoltă de obicei boli pulmonare, iar cei cu varianta 7T sau 9T au un fenotip extrem de variabil, care se poate extinde de la lipsa manifestărilor pulmonare până la forme moderate de boală.
Deoarece mutaţia R117H este asociată cu funcţie pancreatică normală, boala pulmonară mai puţin severă observată la aceste persoane ar putea fi consecinţa stării de nutriţie mai bune6.
Un test genetic negativ pentru mutaţiile ţintite nu poate exclude boala. Deoarece în prezent sunt descrise peste 1000 mutaţii, pe piaţă există mai multe truse de diagnostic, care pot identifica cele mai frecvente mutaţii pentru o anumită zonă geografică sau grup populaţional. Pentru cazurile cu adevărat suspecte se poate apela la metode complexe de analiză genetică a ADN-ului (secvenţiere).
Testele genetice sunt disponibile pentru screening-ul persoanelor asimptomatice care doresc să afle dacă sunt sau nu purtătoare ale genei defective de fibroză chistică şi implică, de obicei, interviuri pre-test, dar şi consiliere privind posibilul impact al rezultatelor testelor pozitive sau negative. Acest tip de analize genetice permite părinţilor să afle dacă au un risc crescut de a avea un copil cu fibroză chistică.
Screening-ul purtătorilor de fibroză chistică se recomandă următoarelor persoane:
- adulţii care au în familie rude cu fibroză chistică;
- partenerii persoanelor cu fibroză chistică; dacă un partener are fibroză chistică şi celălalt este purtător al genei defective de fibroză chistică, atunci copilul va avea 50% şanse de a dezvolta boala;
- cuplurile care doresc să conceapă copii.
Dacă investigaţiile arată că o persoană este purtătoare a genei defective de fibroză chistică, este necesară şi testarea partenerului. Pentru ca un copil să dezvolte afecţiunea, ambii părinţi trebuie să fie purtători ai genei mutante. În cazul în care analizele partenerului sunt negative sunt şanse minime pentru copil să dezvolte boala6.
Specimen recoltat – sânge venos4.
Recipient de recoltare – vacutainer ce conţine EDTA ca anticoagulant4.
Cantitate recoltată – 5 mL sânge4.
Cauze de respingere a probei – folosirea heparinei ca anticoagulant; probe coagulate sau hemolizate4.
Stabilitate probă – 7 zile la 2-8ºC4.
Metodă: PCR şi revers hibridizare pe strip-uri
Testul genetic analizează 38 de mutaţii şi polimorfismul Allele5T-7T-9T în gena CFTR:
F508del, I507del, F508C, I502T, 1706del17, 1677del TA, G542X, 1717-1G>A, R553X, Q552X, G551D, S549R A>C, N1303K, 4016insT, R1162X, R1158X, W1282X, G1244E, 2789+5G>A, 2183AA>G, 711+5G>A, 711+1G>T, G85E, 3849+10kbC>T, 621+1G>T, R117H, D1152H, L1065P, R1066H, L1077P, 4382delA, 1259insA, 852del 22, R347P, T338I, S912X, I148T, 3199del6.
Raportarea şi interpretarea rezultatelor
Vor fi comunicate mutaţiile depistate în gena CFTR şi genotipul respectiv4.
Bibliografie
- Felix A Ratjen MD PhD FRCP(C). Cystic Fibrosis: Pathogenesis and Future Treatment Strategies. In Respir Care 2009 May;54(5):595-605.
- Girish D Sharma. Cystic Fibrosis, www.emedicine.medscape.com, Ref Type: Internet Communication.
- Ioan Popa, Liviu Pop, Zagorca Popa, Casandra Cilt. Ghid de management în mucoviscidoză – (fibroză chistică).
- Laborator Synevo. Referințele specifice tehnologiei de lucru utilizate 2010. Ref Type: Catalog.
- Mayo Clinic/Mayo Medical Laboratories. Test Catalog. Cystic Fibrosis Mutation Analysis, 70-Mutation Panel. www.mayomedicallaboratories.com. Ref Type: Internet Communication
- Samuel M Moskowitz, James F Chmiel, Darci L Sternen, Edith Cheng, and Garry R Cutting.CFTR- Related Disorders. Gene Reviews, 2008. www.ncbi.nlm.nih.gov. Reference Type: Internet Communication.