Pentru detectarea modificărilor cromozomiale asociate cu cancerele hematologice, sursa optimă de celule este măduva osoasă.
Cariotipul din măduva osoasă a devenit în ultimii ani una dintre metodele de elecție în identificarea unor boli hematologice maligne (leucemii, afecțiuni mieloproliferative și limfoproliferative, sindroame mielodisplazice).
CARIOTIPUL DIN MĂDUVA OSOASĂ

- stabilirea intrării în remisiune a bolii
- stabilirea prognosticului bolii
- diagnosticul altor afecțiuni hematologice (sindroame mieloproliferative, afecțiuni limfoproliferative, sindroame mielodisplazice)
- stabilirea terapiei pacienților cu leucemie
- diagnosticul leucemiilor
Dintre afecțiunile hematologice vizibile prin cariotipul din măduva osoasă cele mai frecvent întâlnite în populația generală sunt:
- leucemia mieloidă acută (LMA) (leucemie non-limfocitară acută/leucemie granulocitară acută)
- leucemia mieloidă cronică (LMC)
- sindromul mielodisplazic (SMD)
- leucemiile limfocitare acute (LLA) – cu celule B sau T
- leucemia limfocitară cronică (LLC)
- limfoame (Burkitt, Hodgkin, non-Hodgkin, s.a.)
Modificările cromozomiale observate în urma cariotipului în cazul acestor procese patologice sunt variate, multiple și interesează majoritatea cromozomilor umani: translocații, inversii, duplicații, trisomii, monosomii, deleții, izocromozomi, cromozomi inelari, ș.a.
LEUCEMIA MIELOIDĂ ACUTĂ (LMA)
LMA este caracterizată prin proliferarea anormală a mieloblastilor. Această afecțiune mai este denumită și leucemie mieloblastică acută, leucemie granulomatoasă acută sau leucemie non-limfocitară acută.
Cele mai frecvente modificări cromozomiale asociate acestei patologii sunt observate la nivelul cromozomilor umani 16, 3, 11, 22, 6, 8, 9, 5, 7 (genele ETO/RUNX1, AML1/RUNX1T1, KMT2A/MLL, CBFB, MYH1, MYH11, EV11, PML, RARA, ș.a)..
Detectarea anomaliilor cromozomiale la pacienții cu LMA permite selecția acestora și stabilirea unor grupe de risc în funcție de tipul modificării și cromozomul implicat. Translocațiile între cromozomii 8 și 21 – t(8;21) – și 15 și 17 – t(15;17) și inversiile la nivelul cromozomului 16 – inv(16) – sunt indicatoare ale unui prognostic semnificativ mai bun la persoanele cu LMA. Monosomiile totale sau parțiale ale cromozomilor 5 și 7 (7-, 5-, 7q-, 5q-), precum și modificări la nivelul brațului lung al cromozomului 3 – abn(3q) – sunt corelate evoluției negative a bolii precum și unei durate de viață mai scurte.
LEUCEMIA MIELOIDĂ CRONICĂ (LMC)
LMC a fost prima afecțiune malignă hematologică identificată a fi asociată cu anomalii cromozomiale. Din punct de vedere clinic, LMC este caracterizată prin proliferarea anormală a celulelor granulocitare ca urmare a transformării maligne a unor celule stem pluripotente.
Modificarea cromozomială observată la pacienții cu LMC este translocația reciprocă produsă între brațele lungi ale cromozomilor 9 și 22 – t(9;22)(q34;q11) – cu formarea cromozomului derivativ denumit cromozomul Philadelphia (Ph). Prezența Ph este definitorie pentru diagnosticul de LMC și pacienții cu simptomatologie LMC dar fără Ph ar trebui verificați pentru o altă modificare mieloproliferativă sau sindrom mielodisplazic.
Există variante ale translocației Ph (diferite alte puncte de ruptură sau locații cromozomiale), însă doar la un procent mic de cazuri (5-10%).
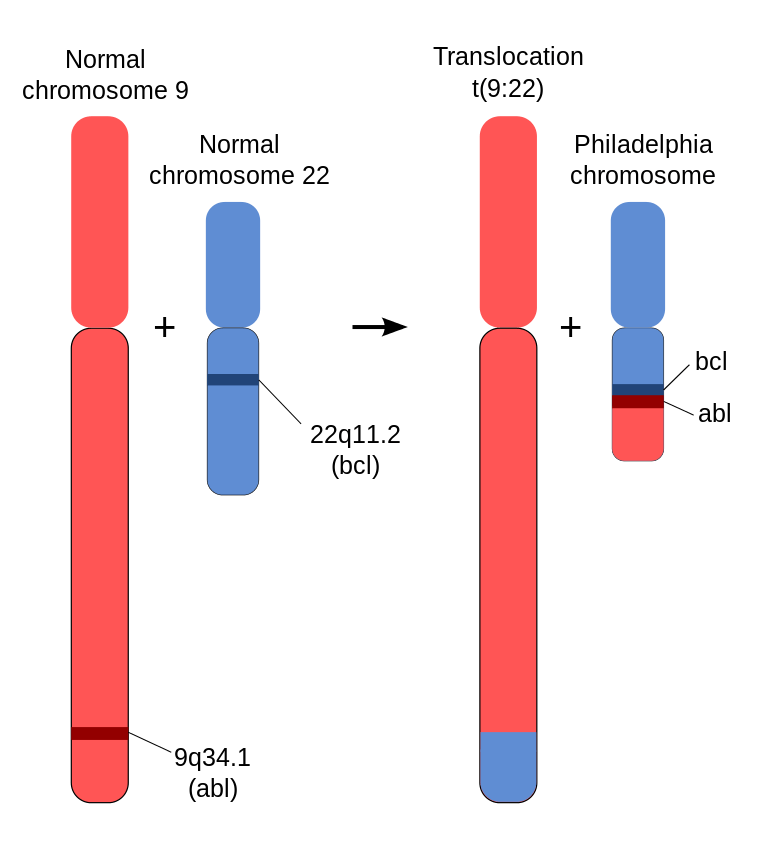
Translocație reciprocă (9;22) – cromozomul Philadelphia
SINDROMUL MIELODISPLAZIC (SMD)
SMD reprezintă un grup de afecțiuni la nivelul celulelor stem hepatopoietice caracterizate prin hematopoeza ineficientă și risc crescut de transformare în LMA.
Pentru confirmarea diagnosticului clinic de SMD, sunt importante analiza unui aspirat din măduva osoasă și analiza cromozomială din măduvă. Eventualele anomalii citogenetice reprezintă un indicator pentru riscul de dezvoltare a LMA.
Printre modificările cromozomiale observate la pacienții cu SMD, cele mai frecvente sunt monosomiile parțiale 5q, 7q, 20q (deleții 5q / 7q / 20q – lipsa brațului lung) sau prezența unor rearanjamente cromozomiale complexe (modificări la nivelul mai multor cromozomi).
LEUCEMIA LIMFOCITARĂ ACUTĂ (LLA)
Transformarea malignă a unor celule stem hematopoietice nediferențiate sau parțial diferențiată determină producerea LLA și LMA.
În cazul LLA, cel puțin 2/3 dintre pacienți prezintă modificări cromozomiale, în timp ce până la 90% dintre copiii diagnosticați evidențiază anomalii citogenetice.
Cele mai frecvente modificări cromozomiale observate la pacienții cu LLA sunt translocațiile reciproce între cromozomii 8 și 21 – t(8;21) – respectiv 15 și 17 – t(15;17) – precum și inversii la nivelul cromozomului 16 – inv(16). Alte anomalii citogenetice sunt monosomia parțială 5q – del(5q), deleții la nivelul brațului lung al cromozomului 9 – del(9p), precum și alte translocații reciproce – t(8;14), t(1;19) (gene de fuziune IGH/MYC, PBX/E2A-TCF3), ș.a.
Translocația t(9;22) (gena de fuziune BCR/ABL) este de asemenea una dintre modificările prezente la pacienții cu LLA.

Gene, locații cromozomiale și modificări genetice în LLA
LEUCEMIA LIMFOCITARĂ CRONICĂ (LLC)
LLC este cea mai frecventă formă de leucemie la populația caucaziană, cu simptomatologie variabilă, de la cazuri neutre până la variante severe și agresive de boală cu evoluție rapidă.
Cea mai frecvent întâlnită modificare genetică la pacienții cu LLC (peste 50% din cazuri) este deleția del(13q14) de la nivelul brațului lung al cromozomului 14, cu afectarea genei RB1.
Trisomia cromozomului 12 este de asemenea una dintre anomaliile genetice comune (10-20% din cazuri). De multe ori trisomia este singura modificare observată la pacienții cu LLC, însă deseori poate fi însoțită și de alte aberații cromozomiale (trisomii 18, 19, deleții 13q, 14q, 11q, 17p, translocații incluzând gena IGH, ș.a.).
Alte anomalii cromozomiale identificate la pacienții cu LLC sunt deleția parțială a brațului scurt al cromozomului 17 – del(17p), cu alterarea genei p53 – precum și translocații continuând gena IGH – t(11;14), t(14;19), gene de fuziune CCND1/IGH și respectiv BCL3/IGH. Inversii la nivelul brațului lung al cromozomului 14 – del(14q23.1) – sunt de asemenea considerate a fi implicate în LLC.
LIMFOAME
LIMFOMUL HODGKIN (LH)
LH este una dintre cele mai frecvente forme de cancer limfatic la nivel mondial, caracterizat printre altele prin hepatosplenomegalie, scădere bruscă în greutate, prurit, stare pre-febrilă, fatigabilitate, dureri lombare, transpirații nocturne, sindrom nefrotic, etc. Până la 50% dintre cazurile de LH sunt produse de virusul Epstein-Barr, existând și o formă imunodeficitară a afecțiunii la pacienții cu HIV/SIDA.
Din punct de vedere molecular există o serie de modificări genice care pot determina producerea LH, cum ar fi mutații la nivelul genelor membre ale căilor de semnalizare NF-KB și JAK/STAT (NFKB1A, NFKB1E, ANFAIP3, SOCS1, REL, JAK2, CD95, TP53.
Din punct de vedere citogenetic, anomaliile cromozomiale observate mai frecvent la pacienții cu LH au fost modificări structurale la nivelul cromozomilor 1 și 11, monosomii parțiale 6q și 7q – del(6q) / del(7q), duplicații la nivelul brațului lung al cromozomului 14 – dup(14q), precum și trisomii ale cromozomilor 3 sau 7. Translocații incluzând gena BCL3 (locație cromozomială 19q13.32), precum și modificări conținând regiunea 4q25-q28 au fost de asemenea, raportate la unii dintre pacienți.
LIMFOAME NON-HODGKIN (LNH)
Limfoamele non-Hodgkin reprezintă un grup de afecțiuni maligne produse de agenți infecțioși, factori de mediu, imunosupresie, inflamație cronică sau rearanjamente cromozomiale.
Anomaliile citogenetice cele mai frecvent întâlnite la pacienții cu LNH sunt modificări structurale care includ regiunea cromozomială 14q32 (gena IGH), cum sunt translocațiile reciproce t(14;18)(q32;q21) și t(8;14)(q24;q32), cu formarea genelor de fuziune BCL2/IGH, respectiv cMYC/IGH. Alte aberații cromozomiale raportate la acești pacienți sunt monosomia parțială 6q – del(6q), izocromozomul 17 – i(17q), trisomii ale cromozomilor 3, 7, 12, 18 sau 21, duplicații la nivelul brațelor scurte ale cromozomilor 2 și 3 – dup(2p), dup(3p), precum și modificări structurale la nivelul cromozomului 17 (ultimele fiind predictive pentru o evoluție agresivă a bolii).
LIMFOMUL BURKITT (LB)
LB este o formă de limfoame non-Hodgkin cu celule B care apare în special la copii și tineri. Dintre cele 3 tipuri de LB (sporadic, endemic și imunodeficitară), tipul sporadic este cel identificat la nivelul populației generale (cel endemic fiind determinat de virusul Epstein-Barr în special la populația africană și cel imunodeficitar fiind prezent la pacienții cu HIV/SIDA). LB este o afecțiune extrem de agresivă care poate afecta sistemul nervos central, măduva osoasă, mezenterul, vezica urinară, rinichii, ovarele și alte organe.
Modificarea citogenetică definitorie pentru LB (identificată în 60-70% dintre cazuri) este translocația reciprocă brațele lungi ale cromozomilor 8 și 14 – t(8;14)(q24;q32), cu formarea genei de fuziune IGH-cMYC – denumită și translocația Burkitt. Această aberație cromozomială este sinonimă cu o evoluție agresivă a afecțiunii.
Alte translocații reciproce care interesează regiunea cromozomială 8q24 (gena cMYC) – t(8;22)(q24;q11) și t(2;8)(p12;q24), gene de fuziune IGL/cMYC, respectiv (IGK/cMYC) – sunt considerații variate ale translocației Burkitt.
La un numar mic de cazuri au fost observate duplicații la nivelul brațului lung al cromozomului 1 (1q21-q25).

Modificări genetice observate la pacienții cu diferite tipuri de afecțiuni hematologoice maligne
PROCEDURA
Specimen recoltat: maduvă osoasă
Etichetarea recipientelor: se face cu etichetă albă, pe care este notat cel puțin barcode-ul.
Transportul probelor: Transportul se efectuează cu respectarea condițiilor de stabilitate a probei: la 20-250C maxim 24 ore din momentul recoltării probei până la intrarea acesteia în lucru.
Condiții de prelucrare și păstrare a probelor biologice până la examinare:
Omogenizarea probei după recoltare și păstrarea până la însămânțare: la 18-250C maxim 24 ore din momentul recoltării probei până la intrarea acesteia în lucru.
Volumul minim necesar: 3 ml maduvă osoasă.
Criterii de respingere a probei: recoltare pe vacutainer incorect; volum insuficient; probe recoltate mai mult de 3 zile; coagulate.
Pregătirea pacientului: pacientul să nu fi urmat tratament cu antibiotic sau să nu fi efectuat radiografii în luna anterioară recoltării.
Prelucrarea necesară după recoltare: omogenizarea probei. Proba se pune în lucru cât mai repede; dacă acest lucru nu este posibil, proba se stochează la 20-25°C maxim 24 de ore.
Recipient de recoltare: recipient cu heparină sodică.